Spongiformní encefalopatie (prionová onemocnění) jsou onemocnění, při nichž se na vývoji podílejí patologické formy prionových proteinů. Víme stále více o prionových onemocněních, ale klíčové aspekty zůstávají neznámé - medicína v současné době nemá prostředky k vyléčení pacientů z těchto nemocí.
Spongiformní encefalopatie, tj. Prionová onemocnění, se mohou vyvinout během života, zatímco jiné vznikají zděděnými genovými mutacemi přítomnými od narození. V rámci této skupiny existuje několik entit vyskytujících se u lidí, například Creutzfeldt-Jakobova choroba nebo fatální familiární nespavost.
Prionové nemoci byly dlouho velmi záhadné. Na rozdíl od jiných patogenů, jako jsou bakterie, viry nebo houby, neobsahují nukleovou kyselinu - priony jsou tvořeny pouze bílkovinami. Teorii prionových nemocí objevil S. Prusiner, tento objev byl ve vědecké komunitě vysoce ceněn - v roce 1997 získal výzkumný pracovník Nobelovu cenu za medicínu. Ačkoli od narození prionového konceptu uplynulo relativně mnoho let, někteří vědci se stále domnívají, že je neúplný, a dále zkoumají povahu těchto stavů - některé z faktorů odpovědných za spongiformní encefalopatie jsou nyní potvrzeny.
$config[ads_text1] not found
Prionové nemoci: příčiny
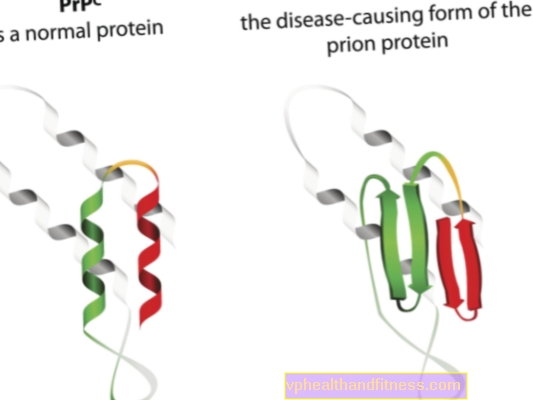
Etiologie prionových onemocnění souvisí s transformací normálních prionových proteinů do patogenních, patogenních forem. Priony jsou molekuly bílkovin, které se nacházejí v těle každého člověka. Jejich funkce ještě není zcela jasná, ale je známo, že za normálních podmínek prionové proteiny nepoškozují tělo. Ale když priony změní svou strukturu a stanou se patogenními částicemi, vyvine se jedna z několika spongiformních encefalopatií. Priony, které se přirozeně vyskytují v těle, se označují jako PRPC, zatímco abnormální formy se označují jako PRPSC. Ty jsou vážným problémem nejen proto, že se mohou hromadit v nervové tkáni ve formě usazenin a způsobit její poškození, ale také proto, že mají schopnost transformovat normální priony do znetvořené formy (jednoduše řečeno PRPSC může „infikovat“ normální proteiny svým patogenním potenciálem).
Přečtěte si také: Huntingtonova choroba (Huntingtonova chorea): příčiny, příznaky, léčba Svalové třesy - příčiny. Co znamená svalový třes? Nemoci, které zabíjejí nejrychleji: SHOCK, EBOLA, DAMN, ATTACK, EMERGENCY [GALE ...V zásadě existují 3 příčiny spongiformní encefalopatie:
$config[ads_text2] not found- sporadické (patogenní mutace se vyskytuje v somatických buňkách, vyskytuje se během života pacienta),
- rodina (vyplývající z břemene mutací zděděných od rodičů),
- pasážované (související se zavedením patogenních prionů do lidského těla, např. prostřednictvím přípravků růstového hormonu kontaminovaných těmito částicemi nebo transplantací rohovky od osoby trpící nějakou spongiformní encefalopatií).
Spongiformní encefalopatie: Creutzfeldt-Jakobova choroba
Creutzfeldt-Jakobova choroba (CJD) byla poprvé popsána na počátku 20. let 20. století. Existují 4 typy onemocnění:
- sporadická CJD (nejběžnější, tvoří až 9/10 všech případů CJD)
- rodné město CJD
- pásový CJD
- varianta CJD
Klinický obraz v průběhu různých variant Creutzfeldt-Jakobovy choroby může být variabilní. Nejběžnější onemocnění v průběhu této skupiny spongiformních encefalopatií jsou:
- poruchy demence (včetně postupného zhoršování paměti, pozornosti a koncentrace)
- myoklonus (mimovolní pohyby jako náhlé trhnutí svalů)
- cerebelární dysfunkce (projevující se například poruchami rovnováhy)
- rozmazané vidění
- pyramidové a extrapyramidové příznaky
V průběhu variant CJD se mohou také objevit duševní poruchy (např. Úzkost, depresivní nálada), bolest a další mimovolní pohyby, které nejsou uvedeny výše.
$config[ads_text3] not found
Prognóza Creutzfeldt-Jakobovy choroby je špatná - například u pacientů se sporadickou CJD to trvá v průměru čtyři až pět měsíců od nástupu symptomů onemocnění až po smrt.
Spongiformní encefalopatie: Gerstmann-Straussler-Scheinkerův syndrom
Gerstmann-Straussler-Scheinkerův syndrom (GSS) obvykle probíhá v rodinách a je způsoben zděděnou mutací genu PRNP. Je považována za nejpomaleji postupující spongiformní encefalopatii. Tým GSS zahrnuje:
- spinocerebelární ataxie
- dysartrie
- poruchy demence
- poruchy polykání
- nystagmus
- zvýšené svalové napětí
Pacienti s diagnostikovanou GSS mají různou dobu a u některých pacientů dochází k úmrtí více než 10 let po nástupu.
Spongiformní encefalopatie: fatální familiární nespavost
Letální familiární nespavost je prionové onemocnění způsobené mutacemi v genu PRNP. Toto onemocnění je extrémně vzácné a bylo dosud diagnostikováno u 28 rodin po celém světě. V průběhu fatální rodinné nespavosti je prvním příznakem neschopnost spát. Tento problém má za následek úzkostné poruchy a pacient má halucinace. Účinkem neustálého nedostatku nočního odpočinku je dysfunkce autonomního systému (včetně změn ve funkci srdce, pocení a poruch trávicího systému), dochází také k postupnému snižování tělesné hmotnosti. V pokročilejších stadiích fatální rodinné nespavosti se objevují hormonální poruchy a v průběhu onemocnění se objevují příznaky demence.
Prognóza fatální familiární nespavosti, stejně jako u jiných spongiformních encefalopatií, je špatná: pacienti obvykle umírají do tří let od jejího vzniku.
Spongiformní encefalopatie: prionopatie s různou náchylností k proteáze
Výskyt těchto spongiformních encefalopatií souvisí hlavně s mutacemi genu PRNP. Tyto mutace se však týkají různých kodonů tohoto genu, a proto se rozlišuje několik různých prionových onemocnění. Relativně nedávno popsanou (v roce 2008) jednotkou je prionopatie s proměnlivou náchylností k proteáze. Lidé trpící touto chorobou nesou mutace až ve třech kodonech genu PRNP.
U prionopatie s různou citlivostí na proteázu se u pacientů vyskytují:
- kognitivní porucha
- extrémní závažnost psychiatrických poruch: mohou to být euforie a agitovanost, ale také významná apatie
- dysartrie
- afázie (poruchy jazyka)
Průměrná doba trvání onemocnění u této prionopatie je méně než 4 roky.
Spongiformní encefalopatie: kuru
Kuru je nyní považován za nemoc, která již prakticky neexistuje - bylo zjištěno u zástupců kmenů z Papuy-Nové Guineje, kteří praktikovali kanibalistické chování. Dominantním příznakem této spongiformní encefalopatie je progresivní cerebelární ataxie. Může být doprovázeno nedobrovolnými pohyby (zejména ve formě chorey, třesu a atetózy) a také močovou a fekální inkontinencí. Pacienti trpící kuru také pociťují výrazné změny nálady, vyvíjejí se u nich primitivní reflexy (např. Sání). Docela charakteristickým problémem v případě této prionové nemoci jsou vynucené záchvaty pláče nebo smíchu - kvůli těmto posledním jevům se kuru někdy říká „smíchová smrt“.
Spongiformní encefalopatie: diagnóza
Prionová onemocnění lze podezřívat na základě pacientových symptomů. Jsou však zcela nespecifické, protože se mohou objevit také v průběhu řady dalších nemocí, které nesouvisejí s priony. Z tohoto důvodu se při diagnostice spongiformních encefalopatií také používají následující:
- zobrazovací testy (např. zobrazování magnetickou rezonancí, které umožňuje detekovat změny související s degenerací mozku prionovými proteiny),
- laboratorní testy (např. stanovení koncentrací proteinů v mozkomíšním moku, např. proteiny MAP-tau, S-100 nebo 14-3-3),
- genetické testy (k detekci přítomnosti mutací u pacienta),
- imunhistochemické testy (s použitím protilátek proti prionovým proteinům).
Diagnózu lze potvrdit také pitvou mozku, ve které je možné najít změny charakteristické pro spongiformní encefalopatie. Mohou to být houbovité léze, různě distribuované a s odlišnou strukturou (v závislosti na konkrétní entitě onemocnění) amyloidové plaky a neuronální defekty.
Spongiformní encefalopatie: léčba
Prionová onemocnění jsou v současné době nevyléčitelná - navzdory četným studiím, které probíhají již mnoho let, medicína stále nemá léky, které by mohly zpomalit nebo úplně potlačit jejich postup. U pacientů se spongiformními encefalopatiemi se používá symptomatická léčba, která je zaměřena na maximální zmírnění intenzity příznaků a zlepšení kvality jejich života.
Práce na léčbě spongiformních encefalopatií však stále pokračují. Vědci se snaží použít různé metody - prvním příkladem je genová terapie. Ovlivnily by nukleové kyseliny a mutace přítomné v jejich struktuře - účelem aplikace genové terapie by bylo neutralizovat chyby v genetickém kódu. Jiný přístup je základem imunoterapie - probíhají práce na vytváření protilátek, jejichž úlohou by bylo eliminovat patogenní priony. Další metodou, která vidí potenciál v boji proti spongiformním encefalopatiím, je léčba pomocí syntetizovaných proteinových molekul, které by po zavedení do těla pacienta neutralizovaly patologické proteiny.
Doporučený článek:
Encefalopatie - příčiny, typy a příznaky





-do-wiczenia-mini-kegla---jak-ich-uywa.jpg)