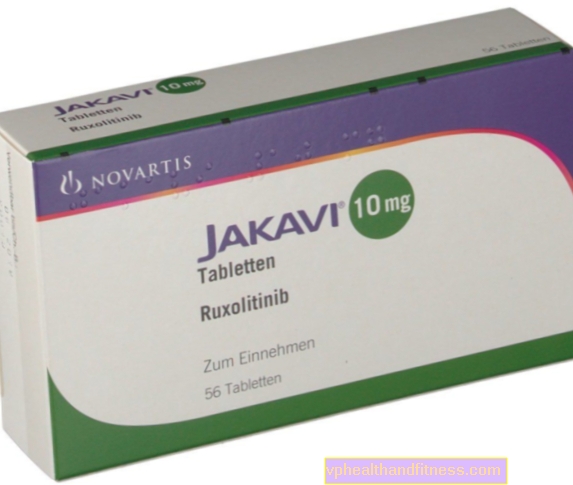
1 tableta obsahuje 5 mg, 10 mg, 15 mg nebo 20 mg ruxolitinibu ve formě fosfátu. Přípravek obsahuje laktózu.
| název | Obsah balení | Léčivá látka | Cena 100% | Naposledy změněno |
| Jakavi | 56 ks, stůl | Ruxolitinib | 2019-04-05 |
Akce
Protinádorové léčivo, inhibitor proteinkinázy. Ruxolitinib je selektivní inhibitor Janusových kináz (JAK), JAK1 a JAK2, které zprostředkovávají signalizaci řady cytokinů a růstových faktorů, které hrají důležitou roli při hemopoéze a imunitní funkci. Vyznačuje se vysokou propustností, dobrou rozpustností a rychlým uvolňováním. Po perorálním podání se rychle vstřebává, Cmax se dosáhne přibližně 1 hodinu po podání. Vazba na plazmatické bílkoviny je přibližně 97%, zejména na albumin. Ruxolitinib neprochází hematoencefalickou bariérou. Je metabolizován hlavně CYP3A4, s dalším přispěním CYP2C9. V plazmě je léčivo přítomno hlavně jako nezměněné léčivo a jako dva aktivní metabolity. Ruxolitinib je vylučován hlavně metabolismem. Průměrná eliminace ruxolitinibu T0,5 je přibližně 3 hodiny, vylučuje se hlavně močí a stolicí.
$config[ads_text1] not found
Dávkování
Orálně. Léčba by měla být zahájena pouze lékařem, který má zkušenosti s podáváním protinádorových léků. Před zahájením léčby je třeba provést kompletní krevní obraz s testem na bílé krvinky. Kompletní krevní obraz s nátěrem bílých krvinek by měl být prováděn každé 2 - 4 týdny, dokud není dávka stabilizována, a poté v závislosti na klinických indikacích. Počáteční dávka. Myelofibróza: 15 mg dvakrát denně u pacientů s počtem trombocytů mezi 100 000 / mm3 a 200 000 / mm3 a 20 mg dvakrát denně u pacientů s počtem trombocytů> 200 000 / mm3. Polycythemia vera: 10 mg perorálně dvakrát denně. Informace o doporučené počáteční dávce u pacientů s počtem trombocytů mezi 50 000 / mm3 a 3 - max. Jsou omezené. Doporučená počáteční dávka u těchto pacientů je 5 mg dvakrát denně a měla by být zvyšována opatrně. Úpravy dávky. Dávky lze upravit na základě bezpečnosti a účinnosti léku. Léčba by měla být přerušena, pokud je počet krevních destiček nižší než 50 000 / mm3 nebo je absolutní počet neutrofilů nižší než 500 / mm3. Léčba by měla být také přerušena u pacientů s PV, pokud jsou hladiny hemoglobinu nižší než 8 g / dl. Po zvýšení počtu krevních obrazů nad tyto hodnoty lze v dávce 5 mg dvakrát denně pokračovat s postupným zvyšováním na základě výsledků úplného krevního testu s nátěrem. Pokud počet trombocytů klesne pod 100 000 / mm3, je třeba zvážit snížení dávky, aby nedošlo k přerušení léčby v důsledku trombocytopenie. Snížení dávky je třeba zvážit také u pacientů s PV, pokud je hemoglobin nižší než 12 g / dl, a snížení dávky se doporučuje, pokud je hemoglobin nižší než 10 g / dl. Pokud je léčba považována za nedostatečně účinnou a krevní obraz je adekvátní, může být dávka zvýšena maximálně o 5 mg dvakrát denně, až na maximum. dávky 25 mg dvakrát denně. Počáteční dávka by neměla být zvyšována během prvních čtyř týdnů léčby a poté by neměla být zvyšována častěji než v dvoutýdenních intervalech. Max. dávka přípravku je 25 mg dvakrát denně. Úpravy dávky při současném užívání silných inhibitorů CYP3A4 nebo flukonazolu. Jednotková dávka by měla být snížena přibližně o 50% a podávána dvakrát denně. Je třeba se vyvarovat současného užívání přípravku s flukonazolem v dávkách vyšších než 200 mg denně. Během léčby silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 se doporučuje častější (např. Dvakrát týdně) monitorování hematologických parametrů a známek a příznaků nežádoucích účinků souvisejících s léčivem. Ukončení léčby. Léčba by měla pokračovat, dokud poměr přínosů a rizik zůstává pozitivní, ale měla by být přerušena po 6 měsících, pokud od zahájení léčby nedošlo ke zmenšení velikosti sleziny nebo zlepšení příznaků. Doporučuje se, aby pacienti vykazující určitý stupeň klinického zlepšení ukončili léčbu ruxolitinibem, pokud se u nich vyskytne 40% prodloužení sleziny ve srovnání s výchozí délkou (přibližně ekvivalentní 25% expanzi sleziny) a není pozorováno žádné skutečné zlepšení jejich sleziny. ve vztahu k příznakům spojeným s onemocněním. Zvláštní skupiny pacientů. U pacientů s mírnou nebo středně těžkou poruchou funkce ledvin není nutná žádná zvláštní úprava dávky. U pacientů s těžkou poruchou funkce ledvin (CCr 3 až 200 000 / mm3.) U pacientů s MF s počtem trombocytů> 200 000 / mm3 se doporučuje jednorázová dávka 20 mg nebo 2 dávky 10 mg podané s odstupem 12 hodin. Následné dávky (jednorázové podání nebo 2 dávky 10 mg podané s odstupem 12 hodin) by měly být podány pouze ve dnech hemodialýzy, po každé dialýze. Doporučená počáteční dávka pro hemodialyzované pacienty s ESRD a PV je jedna dávka 10 mg nebo dvě dávky po každé dialýze. 5 mg podávaných ve 12hodinových intervalech po dialýze a pouze v den hemodialýzy Tato doporučení pro dávkování jsou simulována a je třeba pečlivě sledovat jakoukoli úpravu dávky u pacientů s ESRD z hlediska bezpečnosti a účinnosti. U pacientů podstupujících léčbu nejsou k dispozici žádné údaje. peritoneální dialýza nebo kontinuální veno-venózní hemofiltrace U pacientů s jakoukoli poruchou funkce jater je doporučená počáteční dávka založena na a o trombocyty by měly být sníženy přibližně o 50% a podávány dvakrát denně. Následující dávky by měly být upraveny na základě bezpečnosti a účinnosti léku. Pacienti s diagnostikovanou jaterní dysfunkcí během léčby přípravkem by měli podstoupit kompletní krevní test s nátěrem nejméně 1 za 1-2 týdny po dobu prvních 6 týdnů po zahájení léčby a poté po stabilizaci jaterních funkcí a krevních testů - pokud jsou k dispozici. klinické indikace. Dávka může být upravena tak, aby se snížilo riziko cytopenie. Další úpravy dávky u starších pacientů se nedoporučují. Bezpečnost a účinnost u dětí mladších 18 let nebyla stanovena. Nejsou k dispozici žádné údaje. Způsob dávání. Užívejte s jídlem nebo bez jídla. Pokud dojde k vynechání dávky, pacient by neměl užít další dávku, ale měl by užít další předepsanou dávku.
$config[ads_text2] not foundIndikace
Fibróza dřeně. Léčba zvětšení sleziny spojená s onemocněním nebo příznaky pozorovanými u dospělých pacientů s primární fibrózou kostní dřeně (známá také jako chronická idiopatická fibróza kostní dřeně), fibrózou kostní dřeně předcházenou polycytémií (hyperemií) vera nebo fibrózou kostní dřeně předcházenou esenciální trombocytémií. Všívaná všívaná kachna. Léčba dospělých pacientů s polycythemia vera, kteří jsou rezistentní nebo netolerují léčbu hydroxykarbamidem.
Kontraindikace
Přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku. Těhotenství a kojení.
Opatření
Lék může způsobit hematologické vedlejší účinky, včetně trombocytopenie, anémie a neutropenie, proto je před zahájením léčby vyžadován kompletní krevní test s nátěrem bílých krvinek. Léčba by měla být přerušena u pacientů s počtem trombocytů nižším než 50 000 / mm3 nebo absolutním počtem neutrofilů nižším než 500 / mm3. Bylo poznamenáno, že u pacientů s nízkým počtem krevních destiček (3) v době zahájení léčby je pravděpodobnější výskyt trombocytopenie během léčby. Trombocytopenie je obecně reverzibilní a lze ji obvykle zvládnout snížením dávky nebo dočasným vysazením přípravku, i když v závislosti na klinické indikaci může být nutná transfuze trombocytů. U pacientů, u nichž se vyvine anémie, může být zapotřebí transfuze krve, u pacientů s anémií lze zvážit úpravu dávky nebo ukončení léčby. Pacienti s hladinou hemoglobinu pod 10,0 g / dl na začátku léčby mají větší riziko hladin hemoglobinu pod 8,0 g / dl během léčby ve srovnání s pacienty s vyššími výchozími hladinami hemoglobinu, proto u pacientů s výchozími hladinami hemoglobinu. hemoglobinu pod 10,0 g / dl, doporučuje se častější monitorování hematologických parametrů, stejně jako hodnocení známek a příznaků svědčících o nežádoucích účincích spojených s použitím přípravku. Neutropenie (absolutní počet neutrofilů <500 / mm3) byla obecně reverzibilní a byla zvládnutelná dočasným zadržením léku. Kompletní krevní test by měl být prováděn tak často, jak je klinicky indikováno, a podle potřeby upravit dávku. U pacientů léčených přípravkem se vyskytly závažné bakteriální, mykobakteriální, plísňové, virové a jiné oportunní infekce, proto by pacienti měli být vyšetřeni na riziko závažných infekcí. Lékaři by měli pečlivě sledovat pacienty užívající tento přípravek, zda nevykazují známky a příznaky infekcí, a neprodleně zahájit odpovídající léčbu. Léčba by neměla být zahájena, dokud již nedojde k závažné aktivní infekci. U pacientů užívajících lék na myelofibrózu byla hlášena tuberkulóza a pacienti by měli být před zahájením léčby vyšetřeni na aktivní nebo neaktivní (latentní) tuberkulózu podle místních doporučení. Vyšetřování by mělo zahrnovat anamnézu, možné předchozí kontakty s pacienty s tuberkulózou a / nebo vhodné screeningové testy, jako je rentgen plic, tuberkulinový test a / nebo případně testování uvolňování interferonu-y. Předepisující lékaři by měli mít na paměti riziko falešně negativního tuberkulinového kožního testu, zejména u těžce nemocných pacientů nebo pacientů se sníženou imunitou. U pacientů s chronickou infekcí HBV užívajících lék bylo hlášeno zvýšení viru hepatitidy B (titr HBV-DNA) se současným zvýšením ALT a AST nebo bez něj. Účinek léčiva na virovou replikaci u pacientů s chronickou infekcí HBV není znám; chroničtí pacienti s HBV by měli být léčeni a sledováni podle klinických pokynů. Vzhledem k riziku pásového oparu by lékaři měli instruovat pacienty, aby rozpoznali časné příznaky a doporučí léčbu zahájit co nejdříve. Při použití Jakavi k léčbě MF byla hlášena progresivní multifokální leukoencefalopatie (PML), proto by lékaři měli být ostražití, pokud jde o příznaky, které mohou naznačovat PML, které si pacienti nemusí všimnout (např. Kognitivní, neurologické nebo duševní). Pacienti by měli být sledováni, zda se u nich neobjevují nové nebo zhoršující se příznaky, a pokud se takové příznaky objeví, mělo by se zvážit doporučení pacienta k neurologovi nebo zavedení vhodných diagnostických protiopatření. Pokud existuje podezření na PML, měla by být další léčba pozastavena, dokud nebude PML vyloučen. U pacientů léčených ruxolitinibem, většinou u těchto pacientů s anamnézou dlouhodobé léčby hydroxykarbamidem a předchozími NMSC nebo prekancerózními kožními lézemi, byly hlášeny nemelanomové malignity kůže (NMSC) (včetně bazocelulárního karcinomu, spinocelulárního karcinomu a karcinomu Merkelových buněk). U pacientů se zvýšeným rizikem rakoviny kůže se doporučuje pravidelné kožní vyšetření. Léčba přípravkem byla spojena se zvýšením lipidových parametrů, včetně celkového cholesterolu, HDL cholesterolu, LDL cholesterolu a triglyceridů - doporučuje se sledovat hladinu lipidů a léčit dyslipidemii podle klinických pokynů. U pacientů s těžkou poruchou funkce ledvin by měla být počáteční dávka snížena. U pacientů s terminálním stádiem onemocnění ledvin a MF, kteří dostávají hemodialýzu, by počáteční dávka měla být založena na počtu trombocytů; další dávky by měly být podávány až v den hemodialýzy po skončení každé hemodialýzy. Je třeba provést další úpravy dávkování při pečlivém sledování bezpečnosti a účinnosti léku. U pacientů s poruchou funkce ledvin by měla být počáteční dávka snížena přibližně o 50%. Další úpravy dávkování by měly být prováděny na základě bezpečnosti a účinnosti léku. Pokud se má přípravek podávat současně se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP3A4 a CYP2C9 (např. Flukonazol), měla by být jednotková dávka snížena přibližně o 50% a podávána dvakrát denně. Současné užívání cytoredukčních nebo hematopoetických léčivých přípravků s růstovým faktorem a přípravku nebylo studováno. Po přerušení nebo ukončení léčby se příznaky MF mohou vrátit přibližně za 1 týden.Jsou známy případy pacientů, kteří přerušili léčbu přípravkem, u nichž došlo k závažnějším příhodám, zejména u jiných akutních komorbidních onemocnění. Není známo, zda náhlé přerušení léčby přispělo k výskytu těchto příhod. Lze uvažovat o postupném snižování dávky, kromě případů, kdy je nutné náhlé přerušení léčby, i když užitečnost snižování dávky nebyla prokázána. Přípravek obsahuje laktózu - neměl by být používán u pacientů se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy-galaktózy.
$config[ads_text3] not found
Nežádoucí aktivita
U pacientů s polycythemia vera. Velmi časté: infekce močových cest, anémie stupně 3 (3) a stupně 3 (50 000 - 25 000 / mm3) stupně CTCAE, neutropenie stupně 3.(3) a 4 (3) CTCAE, intrakraniální krvácení, gastrointestinální krvácení, jiné krvácení (včetně epistaxe, pooperační krvácení a hematurie), plyn, elevace alaninaminotransferázy CTCAE stupně 3 ( > 5x - 20x ULN). Méně časté: tuberkulóza. U pacientů s myelofibrózou. Velmi časté: CTCAE anémie jakéhokoli stupně, trombocytopenie jakéhokoli stupně CTCAE, krvácení (jakékoli krvácení, včetně intrakraniálního a gastrointestinálního krvácení, podlitiny a jiné krvácení, podlitiny, jiné krvácení (včetně epistaxe, post-procedurální a hematurie), hypercholesterolemie 1. a 2. stupně CTCAE, hypertriglyceridemie 1. stupně CTCAE, závratě, alanin a CTAE stupně 1 alanin a aspartát, zvýšení aminotransferázy, hypertenze Časté: infekce močových cest, herpes zoster, trombocytopenie 3. stupně (50 CTCAE 000 - 25 000 / mm3), přírůstek hmotnosti, zácpa U MF může dojít k recidivě příznaků MF, jako je únava, bolest kostí, horečka, svědění, noční pocení, symptomatické zvětšení sleziny a úbytek hmotnosti. V klinických studiích s MF se celkové skóre skóre symptomů MF postupně vrátilo na výchozí hodnotu do 7 dnů po ukončení léčby.
Těhotenství a kojení
Použití přípravku během těhotenství je kontraindikováno. Ženy ve fertilním věku by měly během léčby používat účinné metody antikoncepce a pokud žena během léčby otěhotní, je třeba provést individuální posouzení poměru rizika a prospěchu s poradenstvím ohledně možného rizika pro plod. Nesmí se používat během kojení, a proto by mělo být kojení na začátku léčby přerušeno.
$config[ads_text4] not foundKomentáře
Nemá žádný nebo má zanedbatelný sedativní účinek. Pacienti, u kterých se po užití přípravku objeví závratě, by se však měli zdržet řízení nebo obsluhy strojů.
Interakce
Interakční studie byly provedeny pouze u dospělých. Ruxolitinib je vylučován metabolizmem katalyzovaným CYP3A4 a CYP2C9, proto přípravky, které inhibují aktivitu těchto enzymů, mohou vést ke zvýšené expozici ruxolitinibu. Při podávání léku se silnými inhibitory CYP3A4 (jako jsou mimo jiné iboceprevir, klarithromycin, indinavir, itrakonazol, ketokonazol, lopinavir / ritonavir, ritonavir, mibefradil, nefazodon, nelfinavir, posakonazol, saquinavir, jedna jednotka telaviaprev 50% a podává se dvakrát denně. Pacienti by měli být pečlivě sledováni (např. Dvakrát týdně) kvůli možné cytopenii a dávka by měla být postupně zvyšována na základě bezpečnosti a účinnosti. Při použití přípravků, které jsou duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. Flukonazol), je třeba zvážit 50% snížení dávky. Je třeba se vyvarovat současného užívání léčiva s flukonazolem v dávkách vyšších než 200 mg denně. Při užívání induktorů CYP3A4 (jako jsou avasimib, karbamazepin, fenobarbital, fenytoin, rifabutin, rifampin (rifampicin), třezalka tečkovaná (Hypericum perforatum), je třeba pacienty pečlivě sledovat a dávku je třeba postupně zvyšovat na základě bezpečnosti a účinnosti. Pokud se ruxolitinib podává současně s mírnými nebo středně silnými inhibitory CYP3A4 (jako jsou ciprofloxacin, erythromycin, amprenavir, atazanavir, diltiazem, cimetidin), nedoporučuje se žádná úprava dávky, avšak při zahájení léčby středně silnými inhibitory by pacienti měli být pečlivě sledováni ohledně možné cytopenie. CYP3A4: Ruxolitinib může inhibovat intestinální P-glykoprotein a protein rezistence na rakovinu prsu (BCRP), což může vést ke zvýšené systémové expozici substrátům těchto transportérů, jako je dabigatranetexilát, cyklosporin, rosuvastatin a potenciálně digoxin. monitorování léčiv (TDM) nebo klinický stav po podání uvedených látek. Je možné, že potenciální inhibice P-gp a BCRP ve střevě bude minimalizována, pokud bude doba mezi podáváními léčiva udržována co nejdéle. Současné užívání hematopoetických růstových faktorů a přípravku nebylo studováno. Není známo, zda inhibice Janus kinas (JAK) Jakavi snižuje účinnost hematopoetických růstových faktorů nebo zda hematopoetické růstové faktory ovlivňují účinnost přípravku. Současné použití cytoredukčních terapií a přípravku nebylo studováno - bezpečnost a účinnost současného podávání těchto léků není známa. Ruxolitinib neinhibuje metabolismus perorálního substrátu CYP3A4 midazolam - proto se neočekává žádné zvýšení expozice substrátům CYP3A4, pokud se tyto léky podávají současně s Jakavi. Přípravek neovlivňuje farmakokinetiku perorální antikoncepce obsahující ethinylestradiol a levonorgestrel, proto se neočekává, že by se při současném užívání ruxolitinibu snížila účinnost antikoncepčních prostředků obsahujících tuto kombinaci.
Přípravek obsahuje látku: ruxolitinib
Úhrada léku: NE















---co-to-jest-jak-dziaa.jpg)